
Neurofibromatosis Type 2 (NF2)
 Like Neurofibromatosis Type 1 (NF1), Neurofibromatosis Type 2 (NF2) is characterized by the development of slow-growing tumors in the nervous system, particularly along the nerves in the ears. These bilateral “acoustic” or “vestibular” schwannomas are hallmarks of NF2 and can lead to deafness. Tumors can also grow along other nerves in the body including cranial, spinal, optic, and peripheral nerves. Individuals with NF2 can also develop cataracts and meningiomas. Thus, other symptoms may include numbness, pain, vision loss, and balance issues. While these tumors are typically benign, they do have the potential to become malignant. NF2 is much less frequent in the population than NF1, affecting 1 in 25,000, and symptoms appear later, during the teen or early adult years.
Like Neurofibromatosis Type 1 (NF1), Neurofibromatosis Type 2 (NF2) is characterized by the development of slow-growing tumors in the nervous system, particularly along the nerves in the ears. These bilateral “acoustic” or “vestibular” schwannomas are hallmarks of NF2 and can lead to deafness. Tumors can also grow along other nerves in the body including cranial, spinal, optic, and peripheral nerves. Individuals with NF2 can also develop cataracts and meningiomas. Thus, other symptoms may include numbness, pain, vision loss, and balance issues. While these tumors are typically benign, they do have the potential to become malignant. NF2 is much less frequent in the population than NF1, affecting 1 in 25,000, and symptoms appear later, during the teen or early adult years.
The NF2 suppressor gene encodes a cytoskeletal protein called Merlin. Merlin regulates cell signaling pathways that are involved in cell proliferation, growth, invasion, and adhesion. Similar to NF1, there is no cure for NF2. Treatment for NF2 typically involves monitoring and surgery, with chemotherapy and radiation therapy recommended in some situations.
Using genetically engineered mouse models, the Le Lab has provided direct genetic evidence that dysregulation of the Hippo pathway can mediate schwannoma formation, and that RAS/MAPK signaling can modify schwannoma development [JCI Insight, 5(20)141514]. These studies provide important insights into the molecular mechanisms underlying schwannomagenesis and offer promising targets for therapeutic strategies.
For more information, visit Neurofibromatosis Northeast, Neurofibromatosis Network, Neurofibromatosis Midwest, Texas Neurofibromatosis Foundation, and Children’s Tumor Foundation.
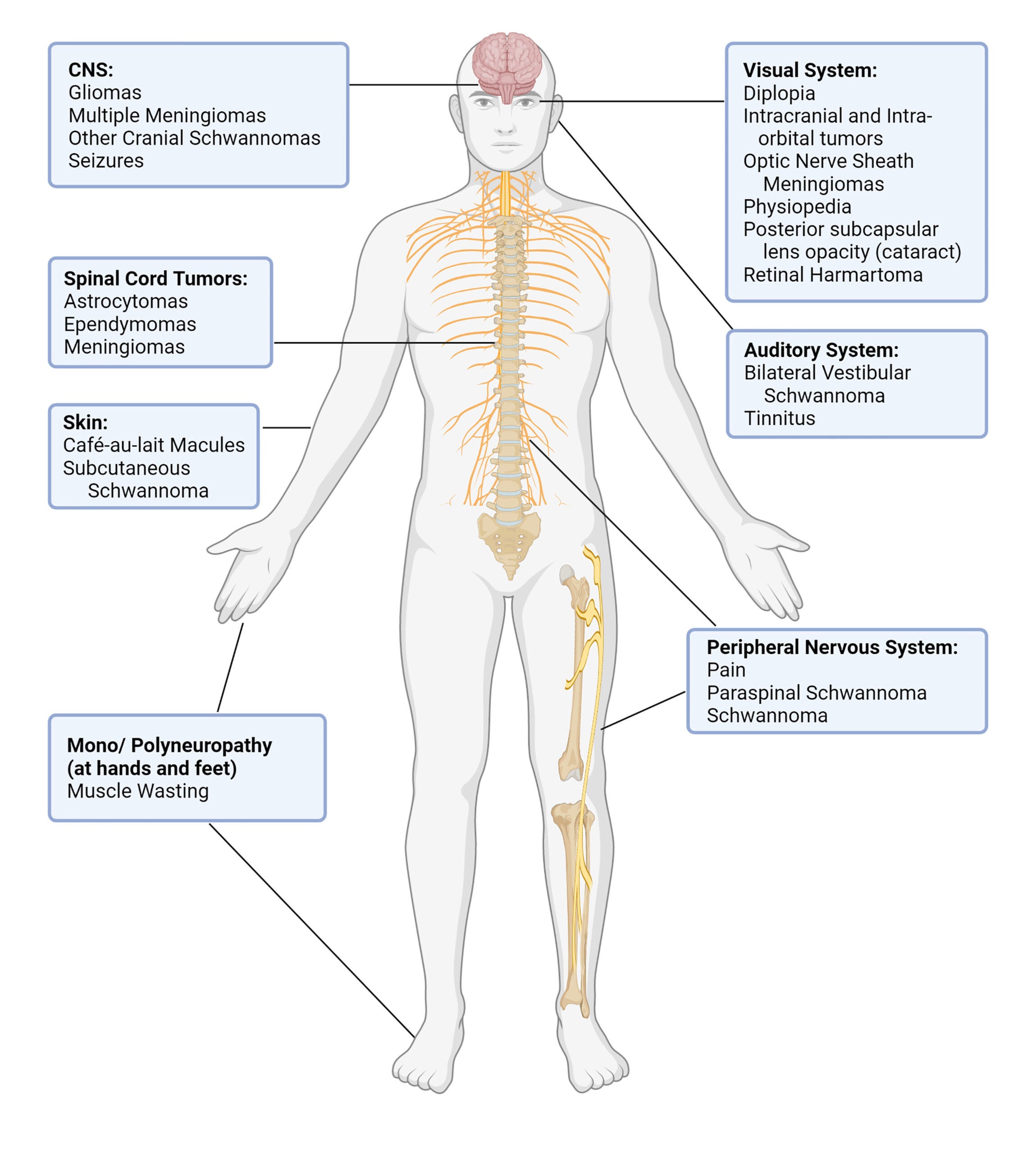
Clinical Manifestations of NF2