
Neurofibromatosis Type 1 (NF1)
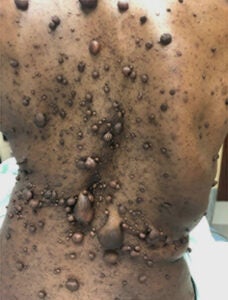
Cutaneous neurofibromas covering the back of an NF1 patient
von Recklinghausen’s Neurofibromatosis Type 1 (NF1) is one of the most common human genetic disorders, affecting 1 in 3,000 live births. NF1 occurs worldwide independent of sex, race, or geographic location. It is caused by mutations in the NF1 tumor suppressor gene, which encodes a GTPase activating protein (GAP) that negatively regulates p21-RAS signaling. Key features of NF1, which typically appear during childhood, are café au lait macules, axillary and groin freckling, and multiple peripheral nerve tumors called neurofibromas. Dermal or cutaneous neurofibromas (cNF) are strictly confined to the skin and do not progress to malignant lesions. cNFs are present in virtually all NF1 patients and can be as numerous as thousands of tumors that cover most of the body surface. Neurofibromas involving the internal nerve plexus are called plexiform neurofibromas (pNF). They are located below the dermis and have an increased risk of malignancy: 8 – 13% of pNFs undergo malignant transformation into malignant peripheral nerve sheath tumors (MPNSTs), which are lethal. Neurofibromas are complex tumors composed mainly of abnormal local cells including Schwann cells, nerves, endothelial cells, fibroblasts, and a large number of infiltrating immune cells. Recent work has demonstrated a critical role for the tumor microenvironment in neurofibroma genesis.
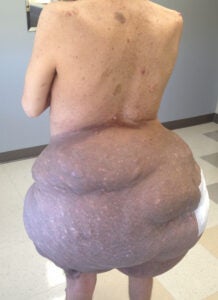
Plexiform neurofibroma on an individual with NF1
Currently, there is no curative treatment for NF1, with surgical removal being the main treatment. Unfortunately, surgery presents a challenge for many patients, some of whom may have thousands of these tumors. Also, surgical procedures are inaccessible to many NF1 patients globally. Therefore, a better understanding of the molecular mechanisms underlying neurofibroma development, as well as the interaction of the tumor cells with the cells in the microenvironment will provide a foothold to develop new therapies aimed at delaying or preventing tumor formation in NF1 patients.
The Le Lab is investigating how early, initiating genetic events such as loss of NF1 interplay with microenvironmental factors to regulate tumorigenesis. While great strides have been made, major knowledge gaps remain in the field including: 1) What drives the oncogenic progression of pNF to MPNST? 2) What is the role of the tumor microenvironment in neurofibromagenesis, and which cell types are involved? 3) Other knowledge gaps in the field pertain to the NF1 gene itself. How does loss of NF1 initiate neurofibroma formation and are certain NF1 mutations associated with certain clinical presentation? These are just a few questions among many regarding NF1 that remain to be answered.
Researchers and clinicians around the world have made important contributions to the NF field, and continue to work to address the knowledge gaps discussed above. A main focus of the lab is to identify the developmental origin or the cell(s) of origin that give rise to NF – a major knowledge gap in the field. The Le Lab developed novel NF models to address this question, including a 3-D in vitro cell culture system, and xenograft and genetically engineered mouse models that recapitulate human neurofibromas. [J Clin Invest, 131(1):e139807; Cancer Cell, 26(5):695-706; Cancer Discovery, 9(1):114-129; Stem Cells, 30(10):2261-2270; Cell Stem Cell, 4(5):453-463]. Using these models, the team was able to identify the cells of origin for pNF and cNF. These studies have also provided important insights into the molecular and cellular pathogenesis of NF and have served as a foothold for development of novel therapies for preventing or delaying tumor formation in NF patients [J Clin Invest, 131(1):e139807; Clin Cancer Res, 25(11):3404-3416; J Clin Invest, 128(7):2848-2861; Nat Communications, 9:5014; Cell Reports, 6(1):81-92; Cell, 152(5):1077-1090; Cancer Research,74(2):586-597; Cancer Research, 71(13):4686-4695].
Video: Surgical removal of cNF
While research is underway to find an effective medical treatment for cNF, which is the greatest burden for individuals with NF1, there is an urgent need to develop a surgical approach that is accessible to most NF1 patients in the world. Dr. Le has developed a robust surgical method to remove cNF that does not require a sterile surgical field, utilizes accessible clinical equipment, and can be performed by any health care provider including family practitioners and physician assistants [JCI Insight, 4(11)e128881:1-9]. Other management strategies for cNF are discussed here: Neuro-Oncology Adv, 2(S1):i107-i116.
For more information, visit Neurofibromatosis Northeast, Neurofibromatosis Network, Neurofibromatosis Midwest, Texas Neurofibromatosis Foundation, and Children’s Tumor Foundation.

Clinical Manifestations of NF1