
May 2024. Two newly minted PhDs! Congratulations to Alison and Sara!
April 2024. James Boehlke joins the lab as a graduate student. James will be working on RNA binding proteins in HSV latent infection
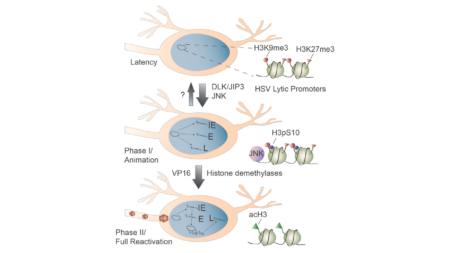
April 2024. Alison successfully defended her PhD on "Heterogeneous Polycomb Silencing of Incoming Herpes Simplex Virus Genomes During Productive Infection". Congrats Alison!
Sara wins the Peach Award for outstanding graduate student in BIMS!
April 2024. The hugestest congratulations to Sara Dochnal for being award the UVA BIMS Michael J. Peach Outstanding Graduate Student award! This award is given to a graduate student who…
Our paper on the targeting of incoming HSV-1 genomes by Polycomb silencing mechanism was published in mBio. Congratulation to first author Allison Francois. This study uses a new method to…