
Research
Herpes simplex viruses type 1 and type 2
Herpes simplex viruses type 1 and type 2 are significant human pathogens. Approximately 67% of the US population are infected with HSV-1 and 20% with HSV-2. The viruses persist in the body for life in the form of a latent, or silent, infection of neurons. Periodically, the viruses reactivate from latency to allow transmission to a new host, which can be associated with disease including cold sores, kerato-conjunctivitis, genital lesions and encephalitis. The Cliffe lab investigates the mechanisms of herpes simplex virus (HSV) latency and reactivation in neurons. We use primary and differentiated neurons along with in vivo models to determine how HSV establishes a latent infection in neurons and how the virus reactivates under conditions of cell stress. Using HSV as a model system, we also aim to understand how different cell types recognize foreign DNA, the role of heterochromatin-associated proteins in gene silencing and the intersection between innate-immune responses and chromatin response to viral infection.
Some of the current projects in the lab include:
Restriction of HSV gene expression in neurons
The HSV genome enters cells with no existing epigentic code. However, in different cell types, different epigenetic modifications are deposited on the HSV genome. We aim to understand how different cell type recognize incoming HSV genomes, how different types of chromatin are deposited onto the HSV genome and how these different chromatin types regulate the outcome of infection. Using a combination of in vivo and in vitro HSV latency models, in addition to imaging approaches and epigenetic techniques, we are specifically investigating the following:
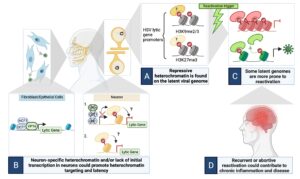
From Whitford & Cliffe, PLoS Pathogens, 2022
- How is heterochromatin targeted to the viral genome in neurons?
- What is the role of restrictive cellular proteins in the establishment of latency?
- How do viral gene products regulate the viral chromatin structure?
- How does the innate immune response intersect with the latent viral genome?
Our long-term aims are to ultimately understand how neurons sense to the foreign, HSV genome and respond by silencing viral gene expression. In the future we also aim to manipulate the viral chromatin structure to make it more repressive and therefore refractory to reactivation.
The mechanisms of HSV reactivation
In response to certain stimuli, latent HSV re-enters the lytic cycle to result in the production of infectious virus that can go on to infect neighboring cells. The silenced viral chromatin forms a barrier for lytic gene expression. Therefore, HSV needs to overcome this barrier for reactivation to occur. We have developed both in vitro and animal models of HSV reactivation that allow us to investigate how the virus overcomes heterochromatin based silencing during reactivation. Using these systems, we are investigating:
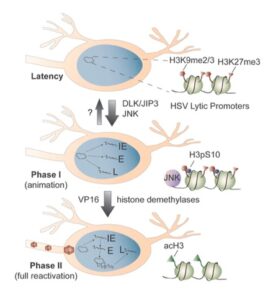
- What are the neuronal stimuli and signaling pathways that can trigger reactivation of the virus?
- How are cellular proteins recruited to the viral genome during reactivation?
- How do cellular pathways intersect with the viral chromatin structure to promote gene expression?
- What determines whether a viral genome undergoes reactivation?
Neuronal responses to HSV infection
HSV persists for life in fully differentiated neurons. As specialized cell types, neurons likely have unique responses to viral infection. The latent HSV genome is nucleosomal, which makes it indistinguishable from host DNA, and is therefore largely hidden from the host immune response. Therefore, it is unknown if and how neurons respond to initial HSV infection and reactivating viruses.
We are investigating the following:
- Do neurons sense and mount innate immune responses to reactivating HSV?
- Certain innate immune pathways restrict reactivation, whereas others are co-opted for reactivation. How do these immune pathways intersect with the viral genome to impact these differential outcomes?
- How do neuronal innate immune responses to HSV change with aging? This is especially important to understand because HSV persists for life and has been implicated in the development of late-onset Alzheimer’s disease.