
Transcriptomics & Long Read Proteogenomics
The transcriptome and the human genome
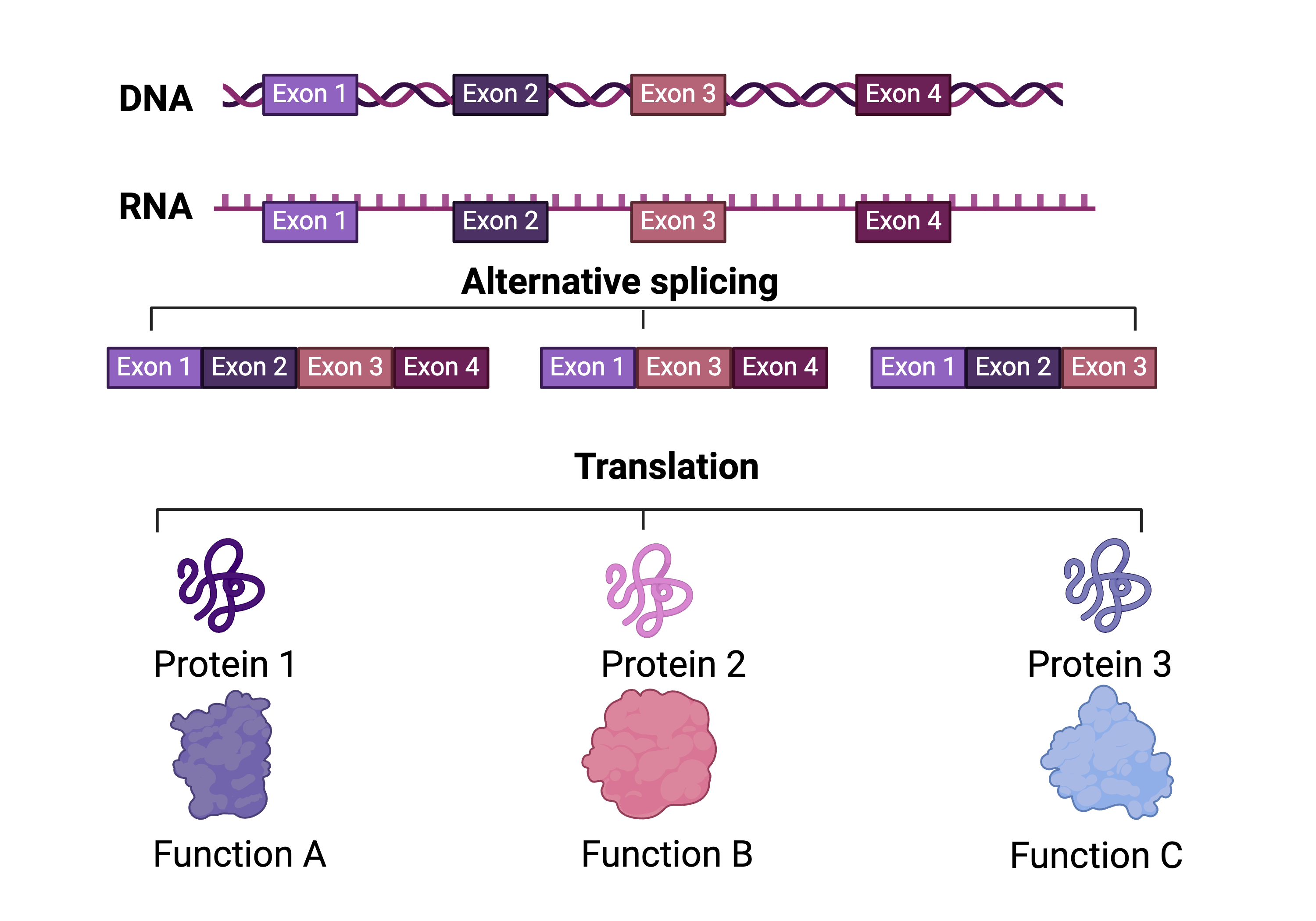
The flow of genetic information within the genome is represented by the central dogma of biology in which DNA encodes genes. These genes become expressed via RNA transcripts and then translated to proteins to form the functional units of the cell. The human transcriptome is quite complex and refers to the comprehensive set of RNA transcripts that are made by the human genome. The content of the transcriptome can vary greatly based off cell type, developmental state or whether the sample represents healthy or diseased conditions. Therefore, the transcriptome provides a measure of a cell status. The process of alternative splicing adds a level of complexity to the transcriptome in which one gene can produce multiple transcripts (or isoforms). These isoforms can go on to produce proteins with potentially distinct functions, therefore contributing to proteomic diversity within the cell. Alternative splicing enables the creation of distinct transcripts by modulating the inclusion or exclusion of stretches of sequence within the RNA. The Sheynkman Lab is very interested in leveraging methods to support understanding of isoforms resulting from alternative splicing to understand how such activity contributes to shaping the transcriptome, and therefore associated proteome.
See the trancsriptome fact sheet on NHGRI’s website for more info!
Capturing isoforms and splicing events
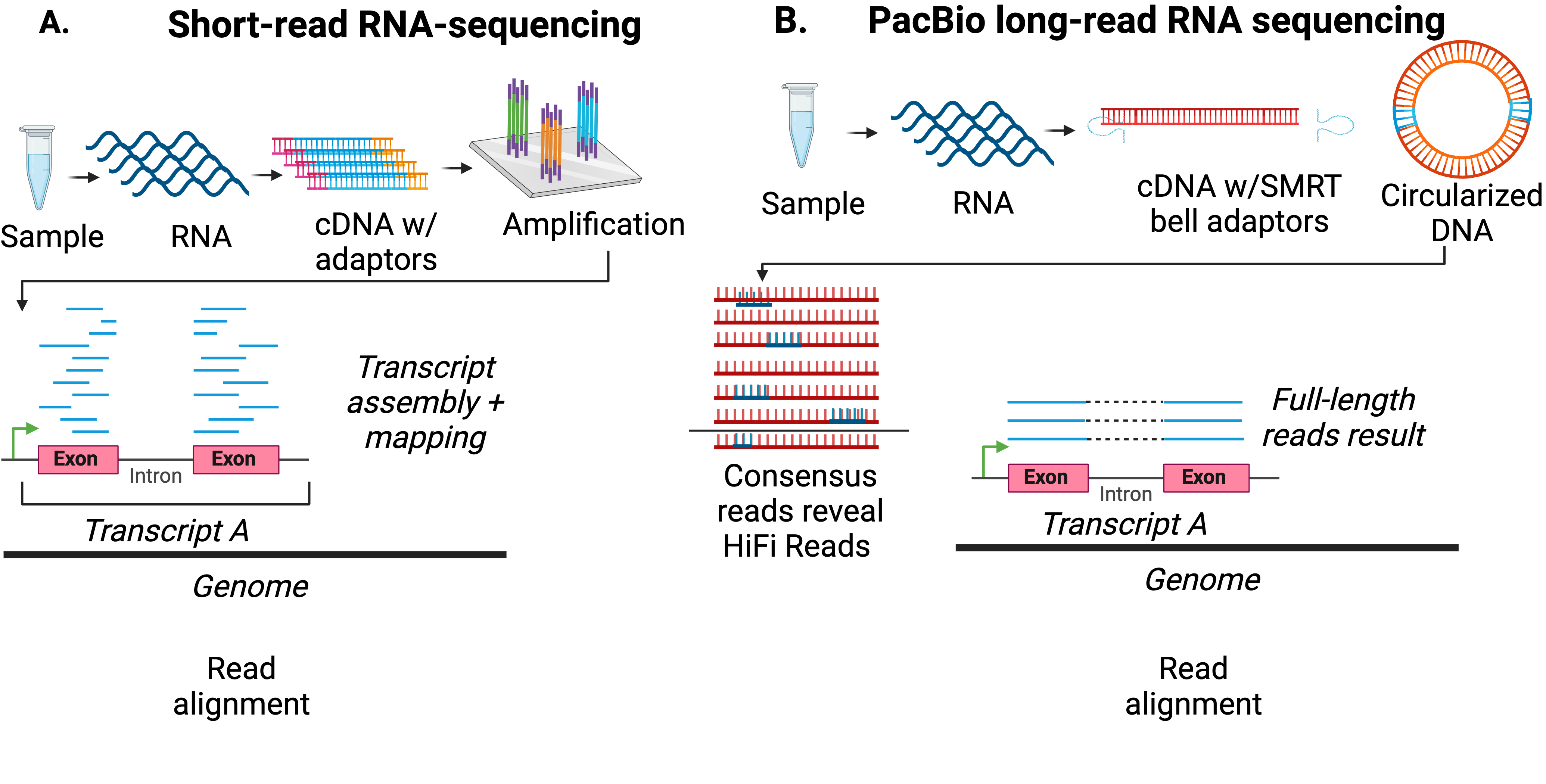
Our understanding of the process of alternative splicing has been informed through the process of sequencing technologies, such as short-read RNA-sequencing technologies such as those offered through Illumina. However, the short-read RNA-sequences that result from such technology often need reconstructed and assembled on a complex genome to determine the transcriptome collected. Isoforms can contain short-stretches of unique sequences and can be masked during transcript reconstruction causing ambiguity of the transcriptome returned, preventing characterization of the distinct isoform produced by alterative splicing. To overcome limitations of short-read RNA-sequencing, we utilize long-read RNA-sequencing as offered through PacBio. This long-read technology results in full-length transcripts allowing for us to characterize isoforms resulting from alternative splicing with confidence in their seqeunces. Many early studies within our lab such as those employed by Miller et al. 2021 and Mehlferber et al. 2022 studied isoform landscapes as they pertain to single samples. Recognizing that the transcriptome is reflective of active cell conditions, recent advances in sequencing technology offered by PacBio through the Kinnex platform have increased yield of sequencing experiments allowing us to obtain confidence in the isoforms collected as we embark on studying cell types in dynamic conditions (ex. development, disease progression etc.). Such new approaches have supported profiling isoforms in dynamic applications to characterize the transcriptome. \
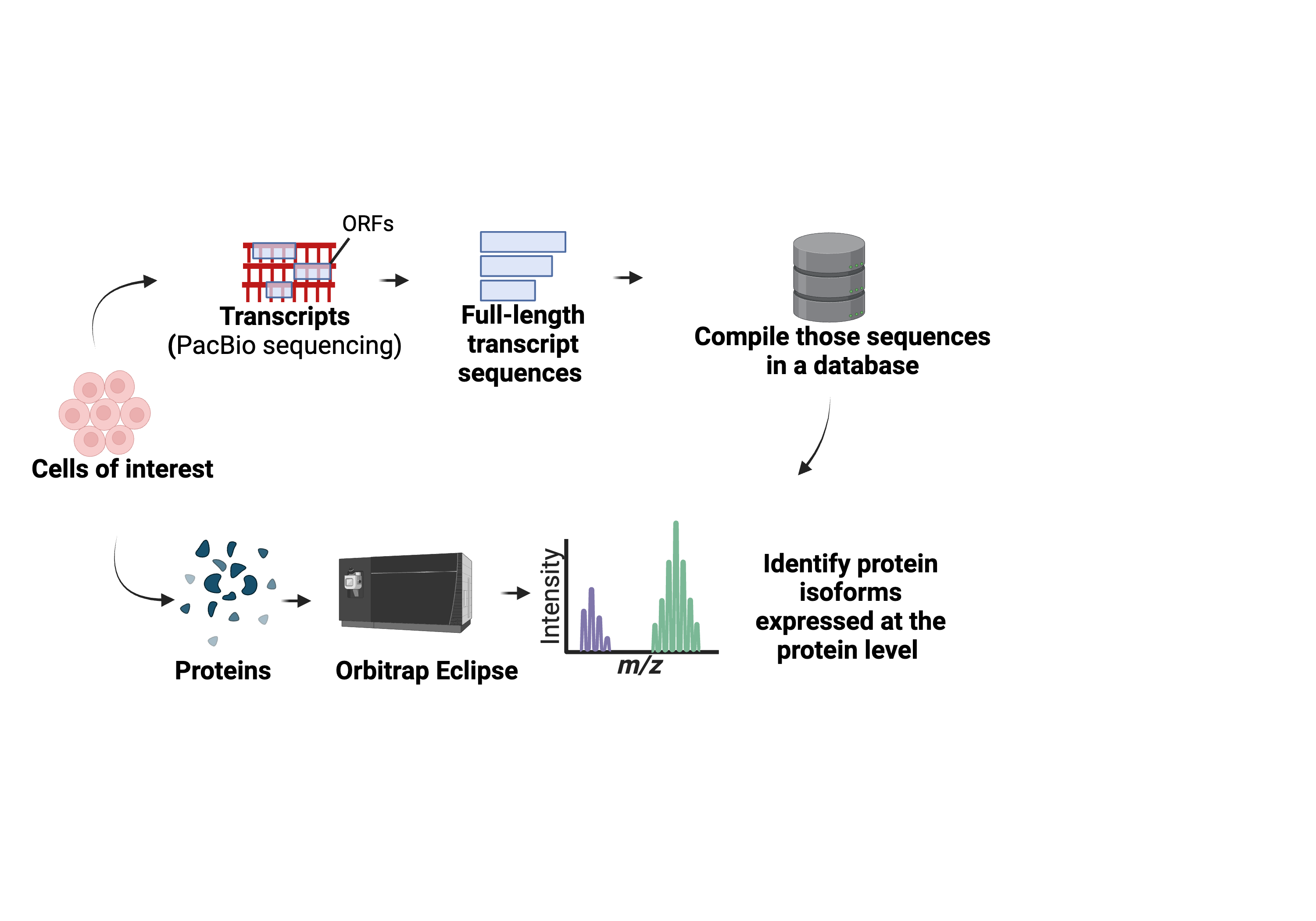
Development of an integrated long-read proteogenomic approach leveraging long-read RNA-sequencing and mass-spectrometry based proteomics to characterize isoforms across the transcriptome
While gaining perspective on the transcriptome provides one level to measure a cell state, understanding the proteome that actively performs the functions within a cell is also critical. To measure the proteins within a sample resulting from the transcriptome, mass-spectrometry-based proteomics remains the gold standard. During mass-spectrometry analysis, peptide sequences are collected and are utilized to map back to a protein sequence and provide protein-level evidence for the existence of a protein. Therefore, for determining proteins expressed in a sample, the content of the sequences within the database are critical to optimize protein identifications. Constructing databases based on the exact transcriptome contents of a cell has been embraced via proteogenomic approaches. Early approaches utilized the transcript collected from short-read RNA-sequencing to compile the database. However, as mentioned, short-read RNA-sequencing can present limitations for isoform identification. Therefore, in 2022, the lab created the first of its kind open-source long-read proteogenomic approach. This approach takes a sample of interest and performs parallel long-read RNA-sequencing and mass-spectrometry based proteomics on the same sample. The transcripts collected from long-read RNA-sequencing are transformed into a database to better represent the transcriptome for the sample. During mass-spectrometry searching, the compiled sample-specific database serves as the resource to determine what proteins and protein isoforms are expressed in a sample. Such approach, also supports potential discovery of novel protein isoforms by mapping back peptides onto these novel stretches of sequence provided by the full-length transcripts, therefore providing protein level evidence for a protein isoform and associated splicing event. This long-read proteogenomic approach has been employed by our lab and beyond to support efforts to better characterize isoforms in various applications.
Some key papers can be found here to learn more on the above discussed approaches:
- Alternative isoform regulation in human tissue transcriptomes, Wang et al. 2008, Nature – one of the first studies describing the prevalence of alternative splicing via RNA-sequencing approaches
- Enhanced protein isoform characterization through long-read proteogenomics, Miller RM, Jordan BT, Mehlferber MM, Jeffery ED, Chatzipantsiou C, Kaur S, Millikin RJ, Dai Y, Tiberi S, Castaldi PJ, Shortreed MR, Luckey CJ, Conesa A, Smith LM, Deslattes Mays A, Sheynkman GM.
- Characterization of protein isoform diversity in human umbilical vein endothelial cells via long-read proteogenomics, Mehlferber MM, Jeffery ED, Saquing J, Jordan BT, Sheynkman L, Murali M, Genet G, Acharya BR, Hirschi KK, Sheynkman GM.