Research
Role of JAK2V617F mutation in the pathogenesis of MPN
Myeloproliferative neoplasms (MPN) including polycythemia vera (PV), essential thrombocythemia (ET), and myelofibrosis (MF) are a group of hematopoietic stem cell-derived myeloid malignancies. A somatic V617F mutation in the JAK2 tyrosine kinase (JAK2V617F) has been found in ~95% cases of PV and 50-60% cases of ET and MF. However, it remains unclear how a single JAK2 mutation contributes to three different MPNs. To investigate the role of JAK2V617F in MPNs, we generated an inducible JAK2V617F knock-in mouse. We observed that expression of heterozygous JAK2V617F mutant in hematopoietic progenitors is sufficient to induce a PV disease in mice. We also observed that homozygous JAK2V617F expression accelerates the progression to MF. Furthermore, we showed that JAK2V617F mutation provides clonal advantage to hematopoietic stem cells.
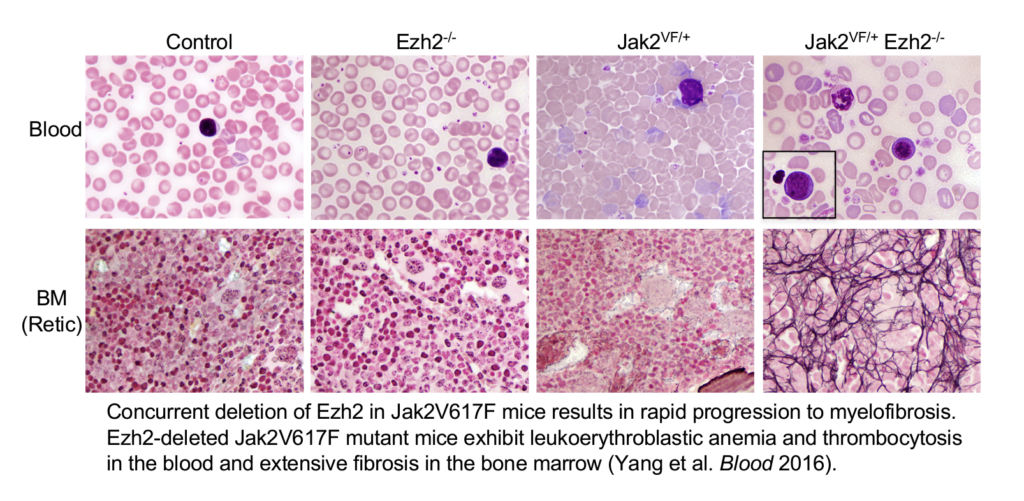
Stat5 is constitutively phosphorylated in hematopoietic cells expressing JAK2V617F. So, we determined the contribution of Stat5 in JAK2V617F-induced MPN. We found that deletion of Stat5 completely blocked the development of PV disease induced by JAK2V617F. Loss-of-function mutations in the histone methyltransferase EZH2 have been found in association with JAK2V617F in patients with MF. So, we assessed the effects of concomitant EZH2 loss and JAK2V617F mutation in MPN using mouse models. We found that loss of EZH2 cooperates with JAK2V617F in the development of myelofibrosis. We also showed that HMGA2 is a downstream target of EZH2 and its expression is elevated in MF. Further, we demonstrated that overexpression of HMGA2 promotes bone marrow fibrosis in JAK2V617F mice.
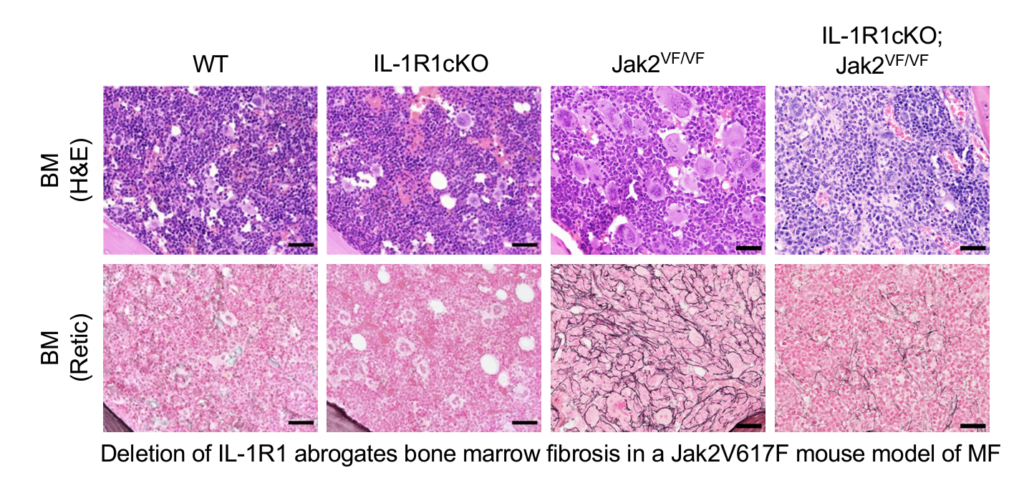
Chronic inflammation is frequently observed in MPNs. Interleukin-1 (IL-1) is considered as a major regulator of inflammation. Recently, we have shown that IL-1 signaling plays an important role in clonal expansion and progression of bone marrow fibrosis in JAK2V617F-induced MPN. We also show that blocking of IL-1R1 using antibodies reduces MPN disease burden and ameliorates bone marrow fibrosis in a JAK2V617F mouse model of MPN.
We are further investigating the contribution of inflammatory signaling, RNA splicing factors and tyrosine phosphatases in the development and progression of MPN induced by JAK2V617F.
Targeted therapies for MPN
Current therapies including JAK2 inhibitors are not sufficient to cure or offer disease remission in patients with MPN/MF. Furthermore, resistance to therapy and transformation to secondary acute myeloid leukemia are observed in many patients after prolonged treatment with JAK2 inhibitors. So, there is a critical need for development of new targeted therapies for MPN/MF.
We have identified CDK6 as a potential therapeutic target in MPN, in particular MF. Expression of CDK6 is significantly elevated in mouse and human MPN hematopoietic progenitors. We investigated the efficacy of CDK4/6 inhibitor Palbociclib alone or in combination with Ruxolitinib in JAK2V617F and MPLW515L murine models of MF. Palbociclib treatment alone significantly reduced leukocytosis and splenomegaly and inhibited bone marrow fibrosis in JAK2V617F and MPLW515L mouse models of MF. Combined treatment of Palbociclib and Ruxolitinib resulted in normalization of peripheral blood leukocyte counts, marked reduction of spleen size, and abrogation of bone marrow fibrosis in murine models of MF. Overall, these data suggest that Palbociclib in combination with Ruxolitinib may have therapeutic potential for treatment of MF and support the clinical investigation of this drug combination in patients with MF.
We also found that PIM1 expression is significantly elevated in MPN/MF hematopoietic progenitors. We observed that genetic ablation of PIM1 blocked the development of myelofibrosis induced by JAK2V617F and MPLW515L. Pharmacologic inhibition of PIM1 using TP-3654 significantly reduced leukocytosis and splenomegaly, and attenuated bone marrow fibrosis in JAK2V617F and MPLW515L mouse models of MF. Combined treatment of TP-3654 and Ruxolitinib resulted in greater reduction of spleen size, normalization of blood leukocyte counts and abrogation of bone marrow fibrosis in murine models of MF. Mechanistically, we show that TP-3654 treatment significantly inhibits mTORC1, MYC and TGF-β signaling in JAK2V617F mutant hematopoietic cells and diminishes the expression of collagen and other fibrotic markers in the bone marrow. Collectively, our results suggest that PIM1 plays an important role in the pathogenesis of MF, and inhibition of PIM1 with TP-3654 could be useful for treatment of MF. This work has led to the design of a multicenter Phase 1/2 clinical trial of PIM1 inhibitor TP-3654 in patients with myelofibrosis (NCT04176198). We are investigating additional new therapies for MPN/MF using pre-clinical models.
Molecular basis for myelodysplasia induced by U2AF1 mutations
Mutations in the genes encoding RNA splicing factors (U2AF1, SRSF2, SF3B1 or ZRSR2) are frequently observed in MDS. U2AF1 is involved in the recognition of the 3’ splice site required for recruitment of the U2 snRNP during pre-mRNA splicing. To investigate the role of wild-type U2AF1 in normal hematopoiesis, we have generated a novel conditional U2AF1 knockout mouse. We observed that deletion of U2AF1 results in profound defects in hematopoiesis characterized by pancytopenia, ablation of hematopoietic stem/progenitor cells (HSPC) leading to bone marrow failure and early lethality in mice. U2AF1 deletion impairs HSPC function and repopulation capacity. U2AF1 deletion also causes increased DNA damage and reduced survival in hematopoietic progenitors. RNA sequencing analysis reveals significant alterations in the expression of genes related to HSC maintenance, cell proliferation and DNA damage response-related pathways in U2AF1-deficient HSPC.
U2AF1 mutations have been identified in ~11% cases of MDS. However, the functional roles of U2AF1 mutations in MDS and the mechanism by which U2AF1 mutations contribute to MDS pathogenesis remain unclear. To determine the roles of mutant U2AF1 in MDS, we have generated a novel conditional U2AF1-Q157R knock-in mouse. We have observed that hematopoietic expression of U2AF1-Q157R mutant results in a macrocytic anemia, erythroid dysplasia and expansion of hematopoietic stem cells (HSC) in the bone marrow. We hypothesize that U2AF1 mutations trigger RNA splicing alterations, gene expression changes, DNA damage and replication stress in HSPC leading to aberrant hematopoiesis. We are investigating the effects of U2AF1-Q157R mutation and underlying molecular mechanisms in myelodysplasia. We will determine the biological and molecular basis for synergy between U2AF1 mutations and co-occurring epigenetic regulator mutations in the pathogenesis of MDS. In addition, we will identify and test therapeutic strategies for U2AF1 mutant MDS. Results from these studies will provide new insights into the molecular pathogenesis of MDS and may lead to new therapeutic approach for treatment of MDS.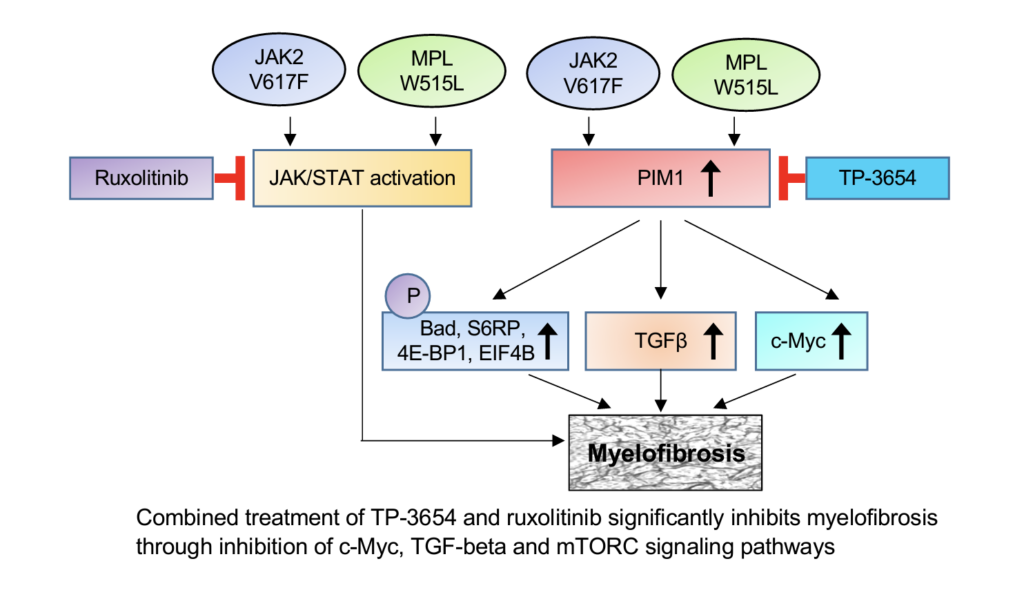
Identification of new therapeutic targets and therapies for triple-negative breast cancer
Triple-negative breast cancer (TNBC) is the most aggressive form of breast cancer and associated with poor prognosis. The vast majority of TNBC-related deaths are due to metastasis to other vital organs. Due to the absence of molecular targets, chemotherapy drugs (e.g., docetaxel, doxorubicin, carboplatin) are commonly used to treat TNBC but these patients eventually develop resistance to chemotherapy. Since current therapies are inadequate to treat TNBC, there is a critical need to identify new therapeutic target(s) and develop novel targeted therapies for metastatic TNBC.
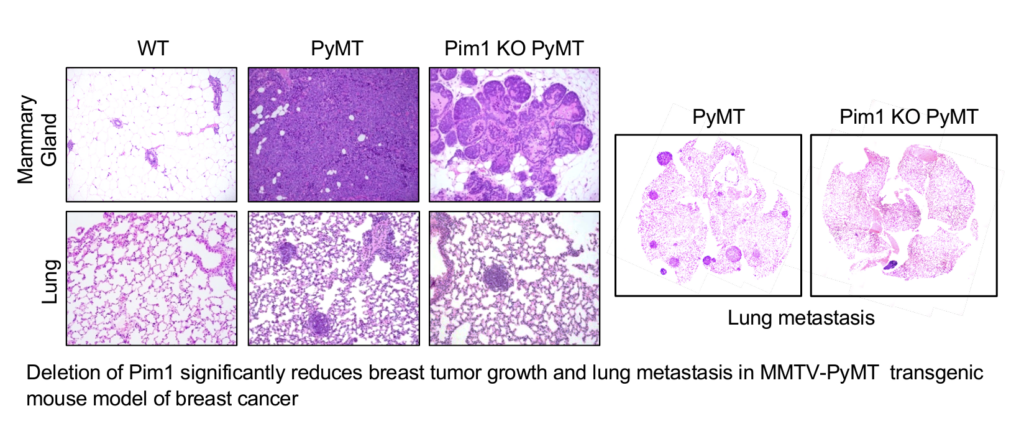
Expression of PIM1 and PIM2 is significantly elevated in TNBC tumors. We have observed that deletion of PIM1 and PIM2 significantly inhibits tumor growth and prevents metastasis in MMTV-PyMT breast cancer model. Treatment with a novel second generation PIM kinase inhibitor TP-3654 significantly reduces the growth and survival of TNBC cells. Moreover, TP-3654 treatment can overcome resistance to chemotherapy in TNBC cells. More importantly, in vivo treatment of TP-3654 markedly inhibits the tumor growth and blocks metastasis in MMTV-PyMT transgenic mouse model of breast cancer. We hypothesize that PIM1 and/or PIM2 play an important role in the progression and metastasis of TNBC, and targeting of PIM kinase using TP-3654 or in combination with chemotherapy might be useful for treatment of TNBC. We will further define the roles of PIM1 and PIM2 in TNBC cell growth, invasion, tumor formation and metastasis. We will test the efficacy of PIM kinase inhibitor TP-3654 alone or in combination with chemotherapy drugs against TNBC cell lines and metastatic breast cancer animal models. We will also identify novel combination therapy that can overcome drug-resistance in TNBC. In addition, we will determine the mechanism by which TP-3654 inhibits TNBC tumor growth and metastasis. Results from these studies will lead to new therapeutic approach for treatment of TNBC.