
Kevin S. Lee, Ph.D.
 Professor
ProfessorResearch Description
Our laboratory is interested in mechanisms of, and treatments for neuronal injury. A multidisciplinary approach has been undertaken to elucidate how neurons are destabilized in these neurological disorders. Knowledge from our basic investigations is then utilized to develop and test specific therapeutic strategies for rescuing damaged neurons. Several lines of investigation are currently underway in the laboratory, including:
- Mechanisms of stroke-related injury and development of novel therapeutic strategies for limiting injury.
- Evaluation of neuroinflammatory mechanisms contributing to persistent cognitive dysfunction after cardiopulmonary bypass surgery. Testing of anti-neuroinflammatory therapeutics for preventing and/or reversing cognitive decline.
- Characterizing of the developmental underpinnings of cortical malformations and identification of physiological mechanisms contributing to seizures.
- Evaluation of neuroinflammatory events contributing to glioblastoma multiforme, which is an aggressive brain tumor that is rapidly and invariably lethal.
Lee Lab Research Projects
Our laboratory is interested in mechanisms of, and treatments for neuronal injury. A multidisciplinary approach has been undertaken to elucidate how neurons are destabilized in neurological disorders. Knowledge from our basic investigations is then utilized to develop and test specific therapeutic strategies for rescuing damaged neurons. Several lines of investigation are currently underway in the laboratory.
1. Persistent cognitive dysfunction is a common complication after cardiopulmonary bypass surgery.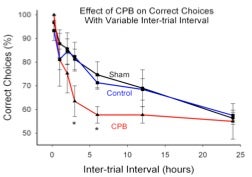 Two central hypotheses for the cause of this disorder are a) the loss of selectively vulnerable neurons due to ischemia, and b) diffuse microinfarcts resulting from a spray of emoboli during CPB. Using a rat model of CPB, we were able to demonstrate impaired performance on a cognitive task in post-CPB animals when tested 6 months after surgery. However, these animals exhibited neither selective neuronal loss nor distributed microinfarction. Rather, the animals exhibited a sustained and localized form of neuroinflammation in which microglial cells were activated in the infragranular zone of the dentate gyrus. In addition, adult neurogenesis was reduced in the same region. Both of these effects persisted for at least 6 months, the longest post-CPB period tested. We hypothesized that the sustained neuroinflammation attenuated adult neurogenesis and that these events contributed to the behavioral impairment after CPB. We tested this concept by blocking microglial activation perioperatively, and then testing behavioral performance 6 months later. Treatment with the minocycline reduced microglial activation, improved adult neurogenesis, and improved behavioral outcomes in CPB animals. Together, these studies support a novel hypothesis for post-CPB cognitive dysfunction, involving neuroinflammation-induced suppression of adult neurogenesis. In addition, the studies provide support for a new therapeutic strategy to limit post-CPB cognitive dysfunction using a drug that is safe and already FDA-approved. Supported by R01 NS050694.
Two central hypotheses for the cause of this disorder are a) the loss of selectively vulnerable neurons due to ischemia, and b) diffuse microinfarcts resulting from a spray of emoboli during CPB. Using a rat model of CPB, we were able to demonstrate impaired performance on a cognitive task in post-CPB animals when tested 6 months after surgery. However, these animals exhibited neither selective neuronal loss nor distributed microinfarction. Rather, the animals exhibited a sustained and localized form of neuroinflammation in which microglial cells were activated in the infragranular zone of the dentate gyrus. In addition, adult neurogenesis was reduced in the same region. Both of these effects persisted for at least 6 months, the longest post-CPB period tested. We hypothesized that the sustained neuroinflammation attenuated adult neurogenesis and that these events contributed to the behavioral impairment after CPB. We tested this concept by blocking microglial activation perioperatively, and then testing behavioral performance 6 months later. Treatment with the minocycline reduced microglial activation, improved adult neurogenesis, and improved behavioral outcomes in CPB animals. Together, these studies support a novel hypothesis for post-CPB cognitive dysfunction, involving neuroinflammation-induced suppression of adult neurogenesis. In addition, the studies provide support for a new therapeutic strategy to limit post-CPB cognitive dysfunction using a drug that is safe and already FDA-approved. Supported by R01 NS050694.
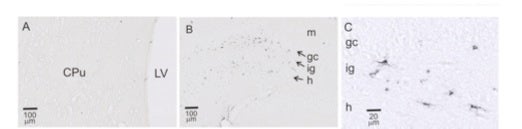
Activated microglial cells are found in the infragranular zone of the dentate gyrus after CPB. This figure shows ED-1-immunopositive (dark appearing), activated microglia in the infragranular zone (ig) of the hippocampus, but not the caudate putamen (CPu). Other abbreviations: LV=lateral ventricle; gc=granule cell layer; h=hilus.
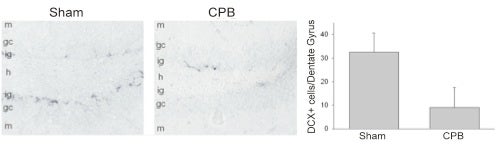
Fewer immature neurons are observed in the infragranular zone of the CPB-treated animals than in Sham-treated control animals. The immunostaining is for doublecortin, which stains for immature neurons.
2. The effective treatment of human stroke remains a critical biomedical challenge. Unfortunately, all major Phase III clinical trials targeting specific mechanisms of neural injury have failed.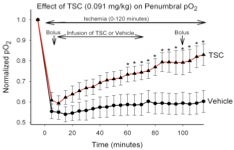 There are several possible explanations for these failures. One compelling explanation is that multiple parallel injury cascades are activated by ischemia, and any one of these cascades may be capable of producing neural damage. Over the last few years, we have undertaken a somewhat different approach toward limiting ischemic injury to the brain. Using a compound that enhances the diffusion of small molecules in aqueous solutions (trans-sodium crocetinate: TSC), we have attempted to improve the supply of metabolic substrates to partially ischemic tissue during an ongoing ‘stroke-like’ event. We demonstrated that tissue oxygenation in an ischemic penumbra can be increased substantially and significantly by TSC. Moreover, TSC reduces the volume of ischemic infarction even when administered 2 hours after the onset of ischemia and under conditions in which the ischemic event was permitted to continue up to 6 or more hours. These rigorous challenges were
There are several possible explanations for these failures. One compelling explanation is that multiple parallel injury cascades are activated by ischemia, and any one of these cascades may be capable of producing neural damage. Over the last few years, we have undertaken a somewhat different approach toward limiting ischemic injury to the brain. Using a compound that enhances the diffusion of small molecules in aqueous solutions (trans-sodium crocetinate: TSC), we have attempted to improve the supply of metabolic substrates to partially ischemic tissue during an ongoing ‘stroke-like’ event. We demonstrated that tissue oxygenation in an ischemic penumbra can be increased substantially and significantly by TSC. Moreover, TSC reduces the volume of ischemic infarction even when administered 2 hours after the onset of ischemia and under conditions in which the ischemic event was permitted to continue up to 6 or more hours. These rigorous challenges were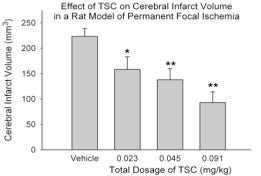 employed in order to better model the conditions observed in human stroke patients. We term this approach “metabolic reflow” because it is effective during the period in which blood flow has not been reestablished. This type of therapy could be value for treating a variety disorders in which ischemia plays a role. Importantly, TSC, which was developed at the University of Virginia, has now passed a Phase I safety trial in humans. (Manabe H, Okonkwo DO, Gainer JL, Clarke R, Lee KS. Protection against focal ischemic injury to the brain by trans-sodium crocetinate. Journal of Neurosurgery 113: 802-809, 2010. Epub:12/09 with podcast; Manabe H, Wang Y, Yoshimura R, Cai Y, Fitzgerald M, Clarke R, and Lee, K.S., Metabolic Reflow as a Therapy for Ischemic Brain Injury, in: Early brain Injury or vasospasm, a crossroad for subarachnoid hemorrhage research and treatment. Hua Feng, Ying Mao, John H. Zhang (eds) Acta Neurochir Suppl. 2010). Supported by R01 NS057168.
employed in order to better model the conditions observed in human stroke patients. We term this approach “metabolic reflow” because it is effective during the period in which blood flow has not been reestablished. This type of therapy could be value for treating a variety disorders in which ischemia plays a role. Importantly, TSC, which was developed at the University of Virginia, has now passed a Phase I safety trial in humans. (Manabe H, Okonkwo DO, Gainer JL, Clarke R, Lee KS. Protection against focal ischemic injury to the brain by trans-sodium crocetinate. Journal of Neurosurgery 113: 802-809, 2010. Epub:12/09 with podcast; Manabe H, Wang Y, Yoshimura R, Cai Y, Fitzgerald M, Clarke R, and Lee, K.S., Metabolic Reflow as a Therapy for Ischemic Brain Injury, in: Early brain Injury or vasospasm, a crossroad for subarachnoid hemorrhage research and treatment. Hua Feng, Ying Mao, John H. Zhang (eds) Acta Neurochir Suppl. 2010). Supported by R01 NS057168.
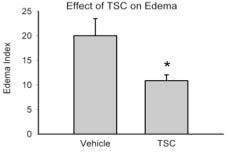
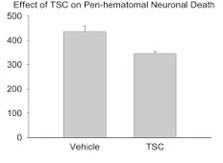
3. A key limiting factor for the rapid treatment of stroke is the need to differentiate between hemorrhagic and occlusive strokes. The only FDA-approved treatment for stroke is tissue plasminogen activator (tPA), which improves outcomes in occlusive strokes but is detrimental in the context of hemorrhagic strokes. The differential diagnosis of hemorrhagic versus occlusive stroke typically takes place in the Emergency Department of a hospital and adds critical time to the evolving ischemic injury. It would therefore be of considerable value to have a therapeutic modality that could be administered independent of the type of stroke. It is plausible that TSC could represent such a therapeutic. We have therefore begun to examine the effects of TSC in animal models of hemorrhagic stroke. In a rat model of intracerebral hemorrhage, TSC improved multiple outcomes. Of particular importance, it lowered the amount of edema in the affected lobe and reduced the number of degenerating neurons in the peri-hematomal area. Studies are now underway using an animal model of subarachnoid hemorrhage (SAH), another form of hemorrhagic stroke. If the SAH studies show improvement or at least no aggravation of outcomes, then TSC would be a reasonable candidate for early intervention in stroke, irrespective of the type of stroke that is occurring. It is our hope that early intervention to improve metabolic supply to partially ischemic tissue will substantially improve stroke outcomes and prolong the therapeutic window for vascular reflow using tPA. Supported by R01 NS057168
4. Cortical malformations are commonly associated with intractable epilepsy. The cortical malformation of subcortical band heterotopia (SBH) is an example of such a disorder. The underlying developmental mechanism postulated to be responsible for the formation of SBH is an error in neuronal migration. Using an animal model of SBH discovered in the Lee laboratory (the tish rat), we examined early developmental events leading to SBH formation. Surprisingly, the primary error in neocortical development in these animals is not a migratory error. Rather, misplaced neuronal proliferation is an early primary event. This represents a novel mechanism for the development of certain neocortical malformations associated with seizures. Moreover, our electrophysiological studies demonstrate that it is the normally-positioned adult neurons, rather than the neurons in the SBH, that trigger seizures. (Trotter, S.A., Kapur, J., Anzivino, M.J., and Lee, K.S., GABAergic synaptic inhibition is reduced before seizure onset in a genetic model of cortical malformation. Journal of Neuroscience 26: 10756-10767, 2006; Trotter SA, Fitzgerald MP, Kapur J, Lee KS. The Tish Rat: an animal model of cortical malformation in the study of epilepsy. In P.A. Schwartzkroin (Ed.), Encyclopedia of Basic Epilepsy Research. pp. 214-219. Oxford: Elsevier (2009); Fitzgerald MP, Covio M, and Lee KS. Disturbances in progenitor cell placement, proliferation, and apoptosis modify neocortical development in the tish neurological mutant. Neuroscience 2010, in press).
5. Glioblastoma multiforme (GBM) is an aggressive brain tumor that is rapidly and invariably lethal. Our laboratory has recently begun to study the growth and invasiveness of glioblastoma. One hypothesis for the high level of invasive activity in GBM is that microglia in the surrounding tissue become activated and through a set of bi-directional signaling pathways facilitate the invasion of glioma cells into the surrounding brain. Minocycline, which as mentioned earlier can suppress microglial activation, has been used effectively in some experimental studies to limit GBM expansion. However, either local administration of minocyline into the glioma or very high levels of systemic minocycline was needed to be effective. A key limitation to this approach is that sustained or recurrent treatment will likely be required using high levels of minocyline. Inasmuch as minocycline is also a potent antibiotic, this type of approach can create multiple complications associated with prolonged antibiotic treatment. We have therefore taken advantage of recently-developed compounds, in which a minocycline derivatives were engineered to be devoid of antibiotic activity while retaining its anti-neuroinflammatory actions. Studies are underway using a syngeneic mouse model of GBM to test the effects of such compounds on tumor expansion.